
Please note: The following information for distributors has been prepared on the basis of the German market and applies exclusively within the EU. If applicable, there are additional, country-specific regulations to be observed which cannot be covered here.
The transition period of the new Medical Devices Regulation 2017/745 (MDR) ends on 26 May 2021. In addition to many new requirements and obligations for us as manufacturers of medical devices, this also contains certain obligations that you as a distributor must fulfil. In order to support you in the implementation of these requirements, we have compiled on this page the most important information on handling our products. This information is specifically geared to our products, which belong to class I and are not sterile. When handling products from other manufacturers, there may be different obligations. Therefore, you will find the complete text of the MDR as well as two guidelines of the DGHIV linked on the right.
Important: For all products you received before May 26th 2021, there is a legally valid sell-by period up to and including 26.05.2025, provided that the shelf life of our products allows this. For these products, the issued declarations of conformity according to the Medical Device Directive (93/42/EEC) are also still valid. The following regulations apply to all products received after May 26th 2021. For these products, we will draw up an EU declaration of conformity in accordance with the Medical Devices Regulation 2017/745 (MDR).
Your duties as a distributor: Duty of care - Duty of inspection - Duty to inform
This information is based on Article 14 of the MDR. In cooperation with the manufacturer, these distributor obligations are intended to ensure that only legally compliant and safe medical devices are supplied to the end users.
Your obligations as a distributor can be divided into three types. The duty of care states that you as a specialist distributor must observe all applicable rules of the MDR with due diligence from 26 May 2021. The duty of inspection and information each includes different tasks, which we explain in more detail below. Some must be completed before dispensing to the patient, others relate to the time afterwards.
Links and Downloads
Information sheets and check list:
MDR information sheet for Ofa distributors
Checklist for inspection obligations
Declarations of conformity:
Download area (Filter Type: Certificates)
Instructions for use:
Instructions for use Support and Compression Stockings
Instructions for use for Supports and Orthoses
The MDR to read:
DGIHV guidelines for implementation:
MDR guide of the DGIHV for manufacturers of custom-made products (German)
1. Before dispensing to the patient: Checking conformity
Before you hand over an Ofa product, you must check a number of things. You should carefully document these tests. You do not need to check every product, but use a sampling procedure that is representative of the products you offer. Ideally, you should do these checks directly at the receiving department before you store the products for final dispensing to the patient.
Before issuing an Ofa product to one of your customers, you must check the compliance of that product with the new MDR directive. To do this, make sure that a declaration of conformity has been issued for the product in accordance with the MDR. For class I products, no notified body is required for this, but Ofa Bamberg issues a declaration of conformity under its sole responsibility. With the declaration of conformity, we declare that the devices covered by it comply with all the requirements of the Medical Devices Regulation 2017/745 (MDR) applicable to these devices. The signed declaration of conformity is also the basis for the CE marking; without a declaration of conformity, this may not be carried out.
You find the Ofa Bamberg declarations of conformity in the download overview on the right.


In addition to the CE mark, you as a distributor must check some other markings and information on our products. You will find these on the packaging, the packaging label or in the instructions for use. What you have to check:
Does the product (packaging/packaging label) bear the following markings?
1. Name or trade name of the product
2. Information from which the user can see what the product is about
3. The name of the manufacturer and the address of his registered place of business
4. An indication that the product is a medical device
5. Information on storage and handling
6. Expiry date (date of manufacture in the case of custom-made products)
As a distributor, you are obliged to comply with the storage and handling instructions as stated on the product labelling. Appropriate evidence of compliance with the storage and handling conditions must be provided.

You will also find the following new label on our packaging:

This shows you at a glance that this product is intended for one-time dispensing to a patient and may be used by exactly this patient several times, according to the instructions for use.
Are instructions for use included?
All Ofa products come with detailed instructions for use. These may also contain assembly instructions for the specialist retailer (usually on the inside cover). With the instructions for use, we fulfil our obligation as a manufacturer of medical products to explain the use of our products and the possible risks during use in a comprehensible way. This includes that in case of distribution abroad, this information is available in the respective official language of the country.
We also make a digital version of our instructions for use available to patients online. You find the links to the instructions for use from the various product areas in the overview at the top right.
Do the instructions for use contain all the information required by the MDR?
This includes:
1. Name of the product
2. Name of the manufacturer and address
3. Date of issue or revision of the instructions for use
4. Information on storage and handling
5. The intended use of the product
6. Information on use, care, indications, contraindications, side effects, residual risks
7. Information on the obligation to notify the manufacturer and the competent authorities in the event of serious incidents

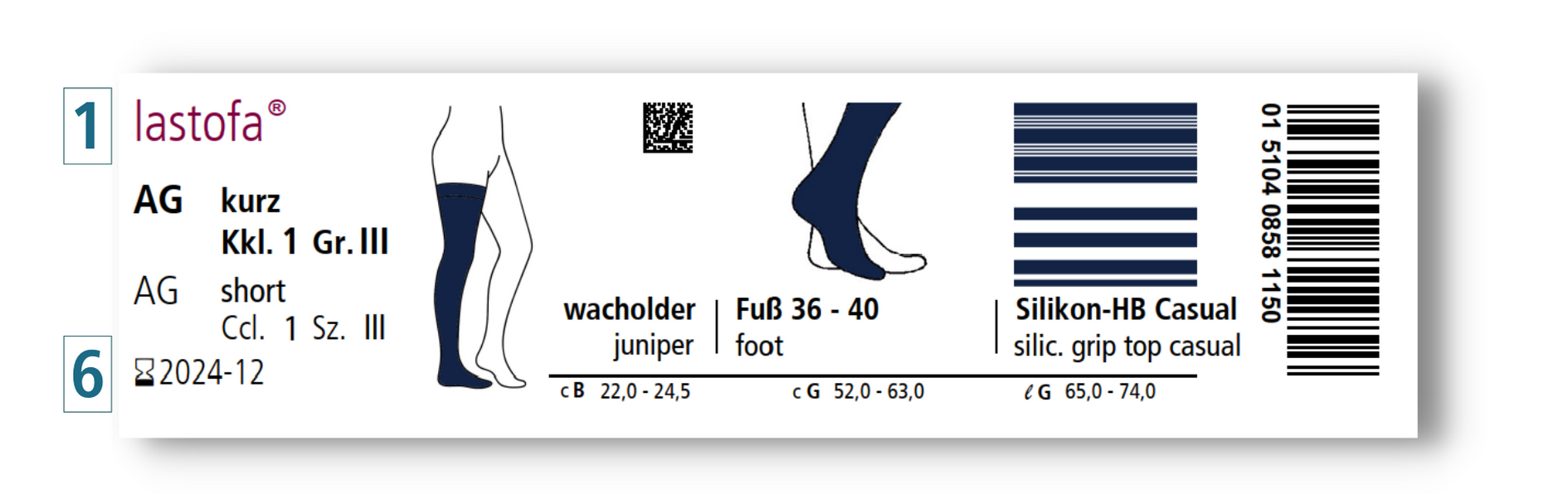
In order to simplify the traceability of products, the so-called Unique Device Identification (UDI) was introduced. Among other things, this consists of a machine-readable barcode or data matrix code that encodes certain information. This information includes the GTIN (Global Trade Identification Number, (01)), which uniquely identifies the product at model level (e.g. Dynamics Carpal Orthosis, size 1, right), as well as the LOT number (10) and the expiry date (17) of the respective individual product. In addition, this information must then also be listed again in legible writing next to the code. The data matrix code and the information in plain writing together make up the complete UDI.
For class I products, this labelling is not mandatory until May 26th 2025. However, we at Ofa Bamberg will already apply the UDI marking this year. The implementation is planned until the end of the year. To give you an idea of what this change will look like, here is a sample label.

Based on the data of the UDI, further information on the products can be found in the central European database "EUDAMED". However, the full functionality of this database will probably not be available until May 2022.
If, after checking the CE marking, declaration of conformity and instructions for use, you come to the conclusion that they do not comply with the requirements of the MDR, you must not release the product to patients. Furthermore, in this case you are obliged to inform us as the manufacturer immediately atvp@ofa.de.
If the product poses a serious risk (see definition below), you as the distributor must also inform the competent higher federal authority. In Germany, this is the Federal Institute for Drugs and Medical Devices (BfArM). You can also find the notification forms online on the BfArM website. You will find more detailed information on what constitutes an incident, a serious incident or a serious public health threat in the next tab.
In addition, you must document the non-compliance of the product. More on this in the tab "Complaint register".
The previous duties of care, such as not giving damaged or defective products to patients, still apply.
2. After delivery to the patient: Follow-up and monitoring
Conformity has been checked, the product has been dispensed. However, this is not the end of your tasks as a distributor. As the first point of contact for patients, you are an important part of the information and reporting chain in case of complaints, technical or application problems and incidents. But what exactly is an incident?
In the course of the MDR, the definition of incident has changed. In addition, the terms serious incident and serious public health threat have been introduced. These terms are defined in article 2 (64.-66.) of the MDR:
Incident
means any malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any undesirable side-effect.
Serious incident
means any incident that directly or indirectly led, might have led or might lead to any of the following:
a) the death of a patient, user or other person,
b) the temporary or permanent serious deterioration of a patient's, user's or other person's state of health,
c) a serious public health threat.
Serious public health threat
means an event which could result in imminent risk of death, serious deterioration in a person's state of health, or serious illness, that may require prompt remedial action, and that may cause significant morbidity or mortality in humans, or that is unusual or unexpected for the given place and time.
As a distributor, you have a duty to cooperate with us as the manufacturer and with the authorities to avert hazards and to enable corrective actions to be taken. If you believe or have reason to believe that a product presents a serious risk, you must immediately inform us as the manufacturer ( vp@ofa.de ) and the competent authorities of the Member States in which you made the product available. In particular, give details of the non-compliance and of any corrective action taken.
As a distributor of medical devices, you are obliged under the MDR to document certain things and to make this evidence available to the manufacturer or the competent authorities on request. In addition to the above-mentioned documentation of random inspections, you must keep a register of complaints.
So if you receive complaints from healthcare professionals, patients or users about suspected incidents related to a product you provide, you must not only forward them immediately to us as the manufacturer (vp@ofa.de) and, if applicable, to the competent authorities, but also record them in a register. In addition to complaints, non-conforming products as well as recalls and withdrawals must also be recorded in this register. The register of complaints must be sent to us as the manufacturer at regular intervals.
Make sure that the documentation is detailed and comprehensible for a third party (e.g. an authority). Authorities may also request samples of the products from you, which you must then provide free of charge.
To ensure the traceability of products, you must be able to provide the following information to the authorities for at least 10 years:
- all economic operators (manufacturers, distributors, importers) from whom you have obtained a product directly
- all economic operators (other distributors, not patients!) to whom you have supplied a product directly
- all health care facilities or health care professionals to whom you have directly supplied a product.
In addition, as a distributor, you are obliged to cooperate with us as the manufacturer as well as with the competent authorities if corrective action or product recalls are necessary.
You might also be interested in this:
Advertising of medical products
As a distributor, you may only use illustrations and information in advertising and information about products that are intended by us as the manufacturer for the respective product. Deviations such as old or other versions, which in any way could lead to a false impression being created regarding the product and its properties, are prohibited.
Please also take into account that you do not give any information verbally or in written form to patients that can be considered as "misleading information".
This includes, but is not limited to, information that
- attribute functions and properties to the product that it does not possess
- give a false impression regarding the treatment or diagnosis and the functions or properties of the product
- fail to inform the user or patient of the anticipated risks associated with the use of the device in accordance with its intended purpose
- suggest uses for the device other than those stated to be part of the intended purpose for which the conformity assessment was carried out
You can find current images of our products in our download area. Registration is required to use this area. If you have not yet registered for our partner portal, you can contact us at partnerportal@ofa.de.

